
Introduction
Sitosterolemia, also known as phytosterolemia, is a rare, autosomal recessive lipid metabolism disorder characterized by elevated plasma and tissue sitosterol levels. This condition arises from increased intestinal absorption and reduced biliary excretion of plant sterols. Clinically, sitosterolemia manifests through xanthomas, premature atherosclerosis, and a spectrum of cardiovascular symptoms including angina pectoris, myocardial infarction, and intermittent claudication. Arthritis, arthralgia, and hemolytic abnormalities can also occur. While over 100 genetically confirmed cases have been reported globally, the genetic underpinnings differ geographically. Asian populations predominantly exhibit mutations in the adenosine triphosphate-binding cassette transporter G family member 5 (ABCG5) gene, whereas Caucasian patients more frequently have mutations in the ABCG8 gene. Advancements in genetic diagnostics have facilitated increased recognition and reporting of sitosterolemia, including within China. This case report details the diagnosis and management of two siblings, a brother and sister, presenting with sitosterolemia and prominent multiple xanthomas, managed at the Burn and Plastic Surgery department. This Familially inherited condition highlights the importance of genetic screening and tailored treatment strategies in managing rare lipid disorders.
Case Report
Case 1
The first patient was a 9-year-old male, the elder sibling of Case 2. Crucially, both parents, while carriers of the mutated gene, were asymptomatic, emphasizing the recessive nature of this familially transmitted disorder. He presented to Dalang Hospital of Dongguan (DHD) with masses on his buttocks and extremities, which had been present for four years.
Case History
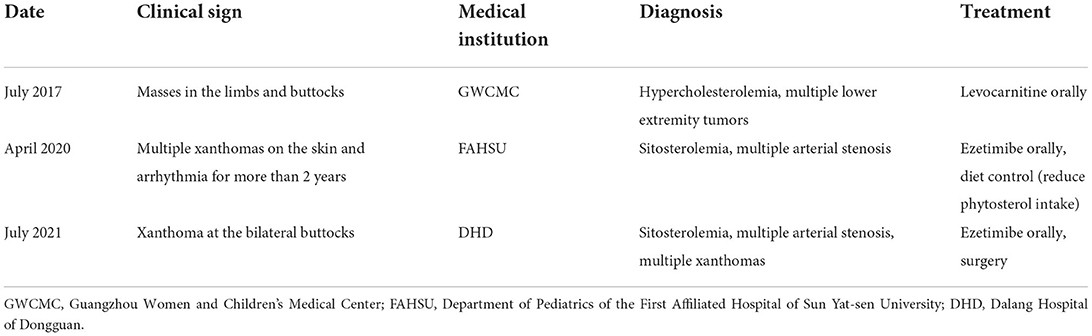
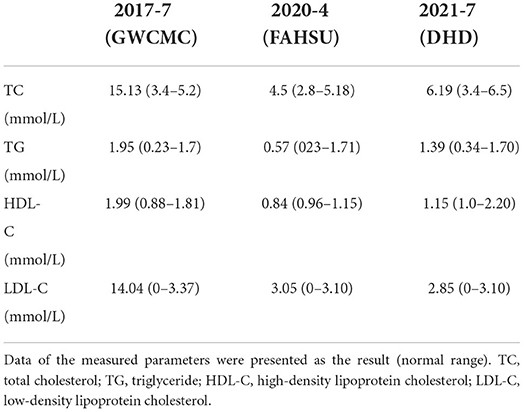
His medical journey, summarized in Table 1, began in July 2017 when he was evaluated at Guangzhou Women and Children’s Medical Center (GWCMC) for “masses in the limbs and buttocks.” Examination revealed multiple xanthomas across his buttocks and limb joints. Initial laboratory findings (Table 2) demonstrated significantly elevated serum total cholesterol (TC) at 15.13 mmol/L, serum triglyceride (TG) at 1.95 mmol/L, high-density lipoprotein cholesterol (HDL-C) at 1.99 mmol/L, low-density lipoprotein cholesterol (LDL-C) at 14.04 mmol/L, apolipoprotein B (Apo B) at 3.08 g/L, apolipoprotein E (APOE) at 95.4 mg/L, and free fatty acids (FFAS) at 1.4 mmol/L. Repeated testing confirmed these elevated lipid parameters. A mass biopsy confirmed xanthoma, and while lipid-lowering treatment was initiated, the xanthomas continued to enlarge.
[Table 1. Basic information of Case 1 from onset to date.
 Basic information timeline of Case 1 from disease onset to diagnosis and treatment, detailing key medical events, diagnoses, and interventions
Basic information timeline of Case 1 from disease onset to diagnosis and treatment, detailing key medical events, diagnoses, and interventions
[Table 2. Blood lipid levels before and after lipid-lowering treatment.
 Comparison of blood lipid levels in Case 1 before and after lipid-lowering treatment, illustrating changes in cholesterol, triglycerides, and lipoproteins
Comparison of blood lipid levels in Case 1 before and after lipid-lowering treatment, illustrating changes in cholesterol, triglycerides, and lipoproteins
In April 2020, he was admitted to the Department of Pediatrics at the First Affiliated Hospital of Sun Yat-sen University (FAHSU) for “multiple skin xanthomas and arrhythmia for over two years.” Physical examination revealed: T=37°C, P=110 bpm, R=24 breaths/min, right upper limb BP=84/65 mmHg, right lower limb BP=102/64 mmHg, left upper limb BP=95/60 mmHg, left lower limb BP=96/48 mmHg, and weight=19.6 kg. He exhibited growth retardation and widespread yellowish-brown, soft nodules, characteristic of xanthomas, particularly prominent around large joints. Xanthelasmas were noted on both upper eyelids. Cardiac examination revealed a heart rate of 110 bpm, an irregular rhythm with premature beats, and strong heart sounds without murmurs. Laboratory results showed: TC=4.5 mmol/L, TG=0.57 mmol/L, HDL-C=0.84 mmol/L, LDL-C=3.05 mmol/L, serum C-reactive protein=41.78 mg/L, erythrocyte sedimentation rate=81 mm/h, interleukin 6=23.26 pg/mL, and albumin=38.2 g/L. Arterial Doppler ultrasonography indicated Takayasu’s arteritis signs, subclavian artery stenosis with partial steal syndrome, bilateral common carotid artery stenosis, abdominal aortic wall thickening and stenosis, and proximal bilateral renal artery narrowing. ECG revealed frequent junctional premature beats, paroxysmal supraventricular tachycardia, and occasional premature ventricular contractions.
Genetic testing for hyperlipidemia, conducted at Guangzhou KingMed Diagnostics Center in April 2020, identified a splice mutation in intron 7 and intron 9 of chromosome 2p21 within the ABCG5 gene. Parental testing confirmed the familial inheritance pattern: the mother carried the heterozygous variant at intron 7, and the father carried the heterozygous variant at intron 9 (Table 3).
[Table 3. Whole-exome sequencing results showing compound heterozygous mutations in the ABCG5 gene for both Case 1 and Case 2.
 Genetic analysis results showing compound heterozygous mutations in ABCG5 gene for both siblings, highlighting specific intron mutations and parental origins
Genetic analysis results showing compound heterozygous mutations in ABCG5 gene for both siblings, highlighting specific intron mutations and parental origins
The final diagnoses were: (1) hereditary sitosterolemia, a familially transmitted condition; (2) multiple arterial stenosis; (3) arrhythmia; and (4) moderate growth retardation. Treatment commenced with ezetimibe for lipid lowering and clopidogrel bisulfate (Talcom) for anticoagulation. While the xanthomas on his buttocks softened, significant shrinkage was not observed.
Examination Results
In July 2021, the patient presented to the outpatient clinic at DHD for xanthoma on his buttocks.
Physical Examination
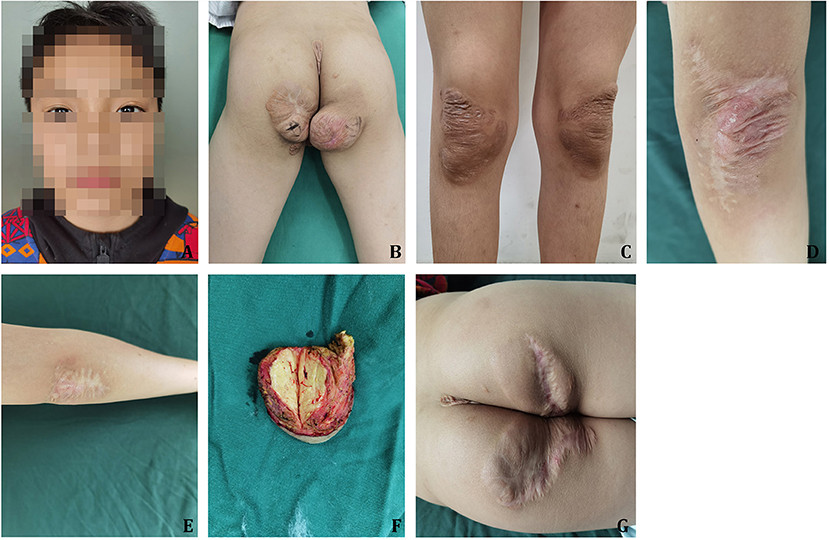
Physical examination revealed: T=36.2°C, P=84 bpm, R=20 breaths/min, and BP=102/53 mmHg. Xanthelasmas were still present on both upper eyelids (Figure 1A). Large, round masses, measuring 7 × 8 cm and 8 × 8 cm respectively, were noted on his buttocks, protruding without ulceration (Figure 1B). Partial shrinkage of knee joint xanthomas was observed following drug treatment (Figure 1C). Surgical scar tissue from previous xanthoma excisions on both elbows was evident (Figures 1D,E).
[Figure 1. Case 1 clinical features and surgical outcomes. (A) Xanthelasmas on the eyelids, a common sign of sitosterolemia. (B) Buttocks xanthoma before surgical removal, demonstrating the size and prominence of the lesions. (C) Partial xanthoma shrinkage in knee joints after lipid-lowering therapy, showing medication effectiveness. (D) Left elbow surgical scar one year post-xanthoma resection, indicating healing and cosmetic outcome. (E) Right elbow surgical scar one year post-xanthoma resection, mirroring the left elbow’s healing. (F) Excised xanthoma tissue during surgery, displaying the characteristic yellowish nodular appearance. (G) Buttocks appearance six months post-xanthoma resection, demonstrating successful surgical removal and healing.
 Clinical images of Case 1 showing xanthelasma, buttocks xanthoma, xanthoma shrinkage with medication, elbow surgery scars, excised xanthoma tissue, and post-surgical outcome
Clinical images of Case 1 showing xanthelasma, buttocks xanthoma, xanthoma shrinkage with medication, elbow surgery scars, excised xanthoma tissue, and post-surgical outcome
Other Examinations
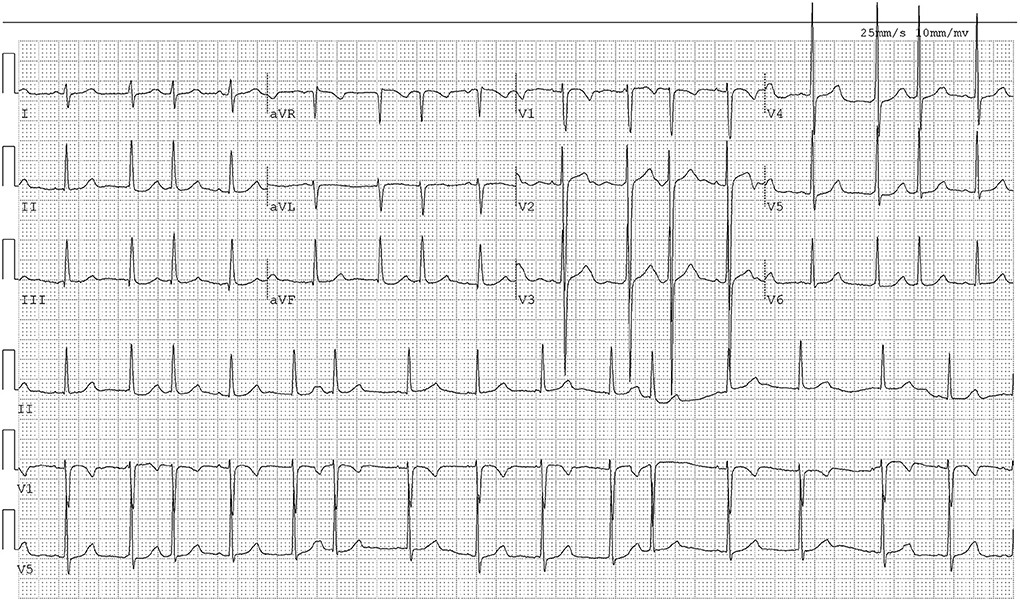
ECG showed sinus arrhythmia and frequent premature atrial contractions (Figure 2). Laboratory tests (Table 2) revealed: TC=6.19 mmol/L, TG=1.39 mmol/L, HDL-C=1.15 mmol/L, and LDL-C=2.85 mmol/L.
[Figure 2. ECG of Case 1 illustrating cardiac arrhythmia. The electrocardiogram tracing shows sinus arrhythmia and frequent premature atrial contractions, indicating cardiac involvement in sitosterolemia.
 Electrocardiogram of Case 1 demonstrating sinus arrhythmia and premature atrial contractions, reflecting cardiovascular complications
Electrocardiogram of Case 1 demonstrating sinus arrhythmia and premature atrial contractions, reflecting cardiovascular complications
Therapeutic Intervention and Outcomes
Surgical resection of the buttocks xanthomas was performed under general anesthesia. The masses were characterized as brownish-yellow nodules, primarily located near joints and pressure points, with distinct boundaries and a soft texture. Incision and dissection revealed the masses to be within the superficial fascia, lacking a capsule, with a pale-yellow, tough cut surface (Figure 1F). Microscopic examination confirmed tuberous xanthoma, composed of foamy histiocytes, multinucleated macrophages, and proliferating fibrous tissue. Postoperative healing was primary and uneventful.
Lipid-lowering and anticoagulation therapy with Ezetimibe and clopidogrel, respectively, was continued post-discharge. A 6-month follow-up showed no xanthoma recurrence (Figure 1G). March 2022 laboratory results were: TG=0.84 mmol/L, TC=6.61 mmol/L, HDL-C=1.52 mmol/L, LDL-C=3.81 mmol/L, and Apo B=1.41 g/L. ECG findings remained consistent.
Case 2
The second patient, the younger sister of Case 1, was a 7-year-old female. She was admitted in January 2022 for “bumps on both elbows for over 2 years.” Her family reported progressively growing masses on her elbows, without pain, ulceration, or movement restriction. She was admitted for surgical treatment of these masses. Like her brother, her condition was familially derived.
Case History
Molecular hereditary testing in March 2020 (Guangzhou KingMed Diagnostics Center) revealed the same splice mutation in intron 7 and intron 9 on chromosome 2p21 of the ABCG5 gene (Table 3) as her brother, confirming the shared familial genetic defect.
Examination Results
Physical Examination
Physical examination showed: T=36.3°C, P=87 bpm, R=20 breaths/min, and BP=105/58 mmHg. She was alert, with normal development and nutrition. Lung sounds were clear, and heart examination was normal except for an irregular rhythm at 87 bpm. No murmurs were auscultated. On the extensor surfaces of both elbows, bulges measuring approximately 7.5 × 7.5 cm (right) and 3.7 × 2.2 cm (left) were observed. The overlying skin was intact, the masses were firm, well-defined, and non-tender, with no joint swelling, pain, or movement restriction (Figure 3A). Xanthelasmas were absent.
[Figure 3. Case 2 xanthoma and post-surgical healing. (A) Elbow xanthomas pre-resection, showing bilateral masses before surgical removal. (B) Right elbow three months post-xanthoma resection, displaying well-healed incision. (C) Left elbow three months post-xanthoma resection, showing successful healing after surgery. (D) Bilateral elbows three months post-surgery, demonstrating symmetrical healing and good cosmetic outcomes.
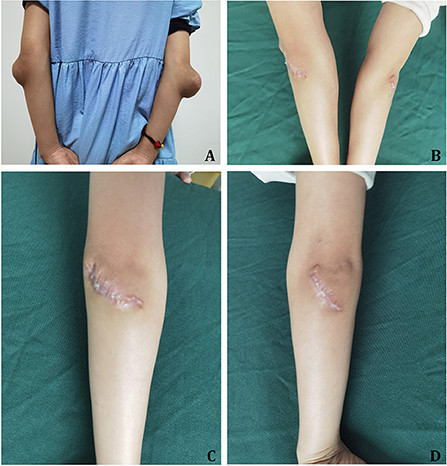 Clinical images of Case 2 showing pre-operative elbow xanthomas and well-healed surgical incisions three months post-resection
Clinical images of Case 2 showing pre-operative elbow xanthomas and well-healed surgical incisions three months post-resection
Other Examinations
Laboratory results included: TG=0.72 mmol/L, TC=7.01 mmol/L, HDL-C=1.65 mmol/L, LDL-C=3.94 mmol/L, HDL-C/LDL-C ratio=0.42, serum Apo A1=1.42 g/L, and serum Apo B=1.57 g/L. ECG showed sinus arrhythmia (Figure 4). Carotid Doppler ultrasound revealed normal internal diameters but irregular thickening of both carotid arteries (1.5 mm and 1.2 mm maximal thickness).
[Figure 4. ECG of Case 2 showing sinus arrhythmia. The electrocardiogram tracing indicates sinus arrhythmia, similar to her brother, suggesting a shared cardiac manifestation of sitosterolemia.
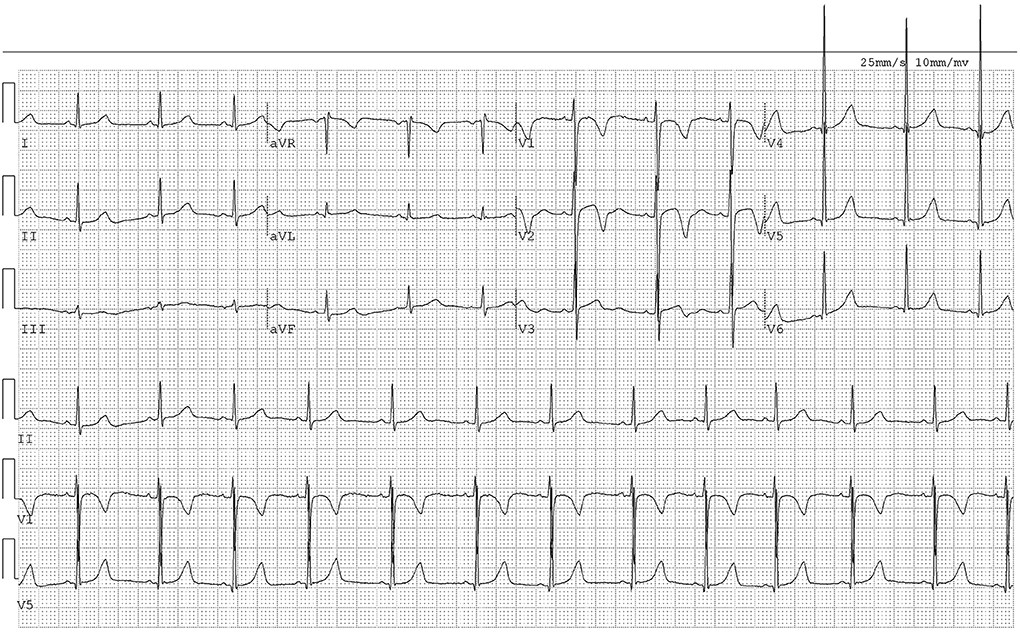 Electrocardiogram of Case 2 demonstrating sinus arrhythmia, highlighting cardiac similarities in affected siblings
Electrocardiogram of Case 2 demonstrating sinus arrhythmia, highlighting cardiac similarities in affected siblings
Therapeutic Intervention and Outcomes
Sitosterolemia was diagnosed, and long-term lipid-lowering therapy with ezetimibe was initiated. Bilateral elbow xanthoma resection was performed, and postoperative pathology confirmed xanthoma.
A 3-month post-operative follow-up showed well-healed incisions (Figures 3B–D). March 2022 laboratory results were: TG=0.54 mmol/L, TC=7.26 mmol/L, HDL-C=1.85 mmol/L, LDL-C=4.22 mmol/L, and Apo B=1.59 g/L. ECG findings remained unchanged.
Importantly, both parents of these pediatric patients had normal cholesterol levels and no signs of xanthomas or atherosclerotic plaques, further emphasizing the recessive familial inheritance pattern.
Results of Hereditary Tests
Test Result Interpretations
Whole-exome sequencing (Table 3) revealed a compound heterozygous mutation consisting of c.1324+1de1G in Intron 7 and Intron 9 of chromosome 2p21 of the ABCG5 gene. The c.904+1G>A mutation was maternally inherited, and the c.1324+1de1G mutation was paternally inherited, confirming the familial genetic basis of the condition. Neither of these specific mutations had been previously reported. Bioinformatics analysis predicted pathogenicity. The ABCG5 variant 2p21 NM_022436.2 Intron 7 p.? c.904+1G>A, a splice mutation, was expected to alter splicing and has been previously reported in sitosterolemia cases and is listed in the dbSNP147 database. The ABCG5 variant 2p21 NM_022436.2 Intron 9 p.? c.1324+1delG, also a splice mutation predicted to affect splicing, was not found in HGMD, ESP6500siv2-ALL, Thousand Genomes, or dbSNP147 databases. Bioinformatics analysis indicated significant impact on mRNA splicing. The presence of a heterozygous pathogenic variant on the trans allele supported its classification as pathogenic, underscoring the familial inheritance and compound heterozygosity in these siblings.
Discussion
Sitosterolemia is an exceedingly rare autosomal recessive disorder of sterol metabolism, often familially inherited, resulting from mutations in genes like LDLR, PCSK9, APOE, and the ABCG gene family. Mutations in ABCG5/ABCG8 disrupt plant sterol metabolism. Berge et al. (2000) linked chromosome 2p21 defects to sitosterolemia. ABCG5/ABCG8 genes are highly expressed in the liver and proximal small intestinal epithelial cells. Phenotype-genotype correlations are weak, with clinical manifestations being largely consistent across different genotypes. The estimated incidence is approximately 1 in 200,000 based on gene frequencies.
The ABCG5/ABCG8 gene products, sterolins, are critical in plant sterol metabolism. Mutations increase intestinal plant sterol absorption and decrease biliary excretion, leading to elevated blood plant sterol levels (sitosterol, campesterol, stigmasterol). Normally, about 50% of dietary cholesterol is absorbed, while plant sterol absorption is minimal due to ABCG5/ABCG8 heterodimeric transport proteins that export free sterols from hepatocytes and intestinal cells into the lumen for excretion, maintaining low plasma plant sterol concentrations. Various ABCG5 gene mutations are documented, with c.1336C>T being most common. However, the c.904+1G>A and c.1324+1de1G mutations in these siblings were novel. Pathogenic variant carriers typically remain unaffected. When both parents are carriers, as in this familial case, there is a 25% risk of offspring inheriting the condition. Other relatives are also at risk of carrying the same pathogenic variant, emphasizing the importance of familial screening and genetic counseling.
Sitosterolemia, a familially transmitted lipid metabolism disorder, often presents in childhood with multiple xanthomas, atherosclerosis, angina, myocardial infarction, hemolysis, anemia, and arthritis. It exhibits a clear familial pattern, and homozygous individuals may have normal or elevated plasma cholesterol, increased LDL, Apo-B, decreased HDL, and Apo-A1. A key diagnostic indicator is significantly elevated plasma phytosterols, particularly sitosterol. In patients with suggestive clinical signs and normal to slightly elevated cholesterol, sitosterolemia should be considered, prompting plasma phytosterol level assessment and familial investigations for diagnostic confirmation.
Sitosterolemia with prominent xanthomas is often misdiagnosed as familial hypercholesterolemia (FH) due to clinical similarities. However, their biochemical, pathogenic, and genetic characteristics differ. FH is autosomal dominant, while sitosterolemia is recessive, highlighting the contrasting familial inheritance patterns. Without early intervention, sitosterolemia prognosis is poor, necessitating accurate differential diagnosis. FH, an autosomal dominant condition linked to the low-density lipoprotein receptor (LDLR) gene, involves cholesterol metabolism. Heterozygous FH mutations lead to premature atherosclerosis and xanthomas in adults, while homozygous mutations cause severe childhood atherosclerosis. FH patients may also experience recurrent arthritis and tenosynovitis without hematologic abnormalities, unlike some sitosterolemia presentations.
Atherosclerosis in sitosterolemia is characterized by early-onset coronary or aortic atherosclerosis, particularly severe in young men. Teenage boys have died from myocardial infarction due to extensive coronary atherosclerosis; one 13-year-old boy with homozygous sitosterolemia and affected siblings succumbed to this complication. A 17-year-old boy with sitosterolemia died suddenly from myocardial infarction during exercise after experiencing progressive angina. Autopsy revealed significant coronary artery occlusion and myocardial infarction. The current cases presented with systemic multiple xanthomas as the primary manifestation. Despite lipid-lowering therapy initiated from disease onset, lack of dietary phytosterol control led to disease progression. Case 1 developed multiple arterial stenoses by April 2020. Case 2, younger and asymptomatic for angina, only showed carotid intima thickening. Post-diagnosis, targeted ezetimibe therapy and dietary phytosterol restriction led to partial xanthoma shrinkage and no significant progression of arterial stenosis by July 2021. This emphasizes the importance of early diagnosis and familial risk assessment for effective management.
Parents’ primary concerns included blood lipid control, xanthoma recurrence post-surgery, and scar formation. Surgical goals were mass removal and restoring normal body contours. Xanthoma locations (buttocks for Case 1, elbows for Case 2) were prone to pressure and friction. Postoperative scarring was minimal, without hypertrophy or pressure ulcers, indicating satisfactory surgical outcomes.
Sitosterolemia, an autosomal recessive disorder, causes both sitosterol and cholesterol elevation. These siblings exhibited hypercholesterolemia, atherosclerosis, and xanthomas. Impaired sitosterol conversion and excretion, and reduced fecal elimination, might expand the body’s sitosterol pool, increasing LDL uptake via LDLR, leading to tissue sitosterol deposition and xanthoma formation. Pediatric sitosterolemia features include skin/tendon xanthomas, arthralgia, hemolytic anemia, thrombocytopenia, splenomegaly, and hyperphytosterolemia. Some children may only present with hematologic abnormalities without xanthomas. Jamwal et al. reported Indian children with stomatocytosis, anemia, giant thrombocytopenia, growth arrest, and no xanthomas, later diagnosed with sitosterolemia due to a homozygous ABCG5 mutation, highlighting the variable clinical presentation of this familially inherited condition.
The primary pathological mechanism involves excessive plant sterol absorption, slow excretion, and insufficient endogenous cholesterol synthesis due to inadequate hydroxymethylglutaryl coenzyme A (HMG-CoA) reductase activity. Thus, reducing sitosterol blood and tissue concentrations requires decreasing absorption and increasing excretion.
Current sitosterolemia treatments include dietary control, lipid-lowering drugs, and intestinal bypass surgery. Ezetimibe, a selective cholesterol absorption inhibitor, blocks exogenous cholesterol absorption by targeting intestinal cholesterol transporters. Strict diet and bile acid sequestrants can partially reduce xanthomas and improve lipid profiles. Surgical xanthoma removal is indicated for function or appearance concerns. Post-surgery ezetimibe may prevent tumor recurrence and intimal lipid plaque accumulation. This comprehensive approach, encompassing dietary, pharmacological, and surgical interventions, is crucial for managing this familially inherited disorder.
Data availability statement
All data generated or analyzed during this study are included in this article. Further enquiries can be directed to the corresponding author.
Ethics statement
Written informed consent was obtained from the minor’s legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
S-QS and D-SX: conception and design of the research. D-SX: acquisition of data and critical revision of the manuscript for intellectual content. S-QS and X-MD: analysis and interpretation of the data. J-AK and Y-CL: statistical analysis. S-QS: writing of the manuscript. All authors read and approved the final draft.
Acknowledgments
We are particularly grateful to all the people who have given us help on our article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
- Bastida JM, Girós ML, Benito R, Janusz K, Hernández-Rivas JM, González-Porras JR. Sitosterolemia: diagnosis, metabolic and hematological abnormalities, cardiovascular disease and management. Curr Med Chem. (2019) 26:6766–75. doi: 10.2174/0929867325666180705145900
PubMed Abstract | CrossRef Full Text | Google Scholar
- Buonuomo PS, Iughetti L, Pisciotta L, Rabacchi C, Papadia F, Bruzzi P, et al. Timely diagnosis of sitosterolemia by next generation sequencing in two children with severe hypercholesterolemia. Atherosclerosis. (2017) 262:71–7. doi: 10.1016/j.atherosclerosis.2017.05.002
PubMed Abstract | CrossRef Full Text | Google Scholar
- Zhuang LL, Yao W, Liu HM, Li GM, Zhang T, Guan WZ, et al. Thalidosteroemia in children with recurrent fever and joint pain. J Southeast Univ. (2021) 40:528–32. doi: 10.3969/j.issn.1671-6264.2021.04.018
CrossRef Full Text | Google Scholar
- Berge KE, Tian H, Graf GA, Yu L, Grishin NV, Schultz J, et al. Accumulation of dietary cholesterol in sitosterolemia caused by mutations in adjacent ABC transporters. Science. (2000) 290:1771–5. doi: 10.1126/science.290.5497.1771
PubMed Abstract | CrossRef Full Text | Google Scholar
- Su X, Shao Y, Lin Y, Zhao X, Zhang W, Jiang M, et al. Clinical features, molecular characteristics, and treatments of a Chinese girl with sitosterolemia: a case report and literature review. J Clin Lipidol. (2019) 13:246–50. doi: 10.1016/j.jacl.2019.01.007
PubMed Abstract | CrossRef Full Text | Google Scholar
- Xu L, Wen W, Yang Y, Xie J, Li R, Wu Y, et al. Features of sitosterolemia in children. Am J Cardiol. (2020) 125:1312–6. doi: 10.1016/j.amjcard.2020.01.048
PubMed Abstract | CrossRef Full Text | Google Scholar
- Tada H, NohaRa A, Inazu A, Sakuma N, Mabuchi H, Kawashiri MA, et al. Sitosterolemia, hyper cholesterolemia, and coronary artery disease. J Atheroscler Thromb. (2018) 25:783–9. doi: 10.5551/jat.RV17024
PubMed Abstract | CrossRef Full Text | Google Scholar
- Patel SB, Graf GA, Temel RE. ABCG5 and ABCG8: more than a defense against xenosterols. J Lipid Res. (2018) 59:1103–13. doi: 10.1194/jlr.R084244
PubMed Abstract | CrossRef Full Text | Google Scholar
- Tada H, Nomura A, Yamagishi M, Kawashiri MA. First case of sitosterolemia caused by double heterozygous mutations in ABCG5 and ABCG8 genes. J Clin Lipidol. (2018) 12:1164–8.e4. doi: 10.1016/j.jacl.2018.06.003
PubMed Abstract | CrossRef Full Text | Google Scholar
- Nguyen LB, Cobb M, Shefer S, Salen G, Ness GC, Tint GS. Regulation of cholesterol biosynthesis in sitosterolemia: effects of lovastatin, cholestyramine, and dietary sterol restriction. J Lipid Res. (1991) 32:1941–8.
PubMed Abstract | Google Scholar
- Feng S, Zhao X, Wang Y, Wang Y, Chen G, Zhang S. Autosomal recessive hypercholesterolemia caused by a novel LDLRAP1 variant and membranous nephropathy in a Chinese girl: a case report. Front Cardiovasc Med. (2022) 9:811317. doi: 10.3389/fcvm.2022.811317
PubMed Abstract | CrossRef Full Text | Google Scholar
- Tada H, Kawashiri MA, Okada H, Endo S, Toyoshima Y, Konno T, et al. A rare coincidence of sitosterolemia and familial mediterranean fever identified by whole exome sequencing. J Atheroscler Thromb. (2016) 23:884–90. doi: 10.5551/jat.34827
PubMed Abstract | CrossRef Full Text | Google Scholar
- Salen G, Shefer S, Nguyen L, Ness GC, Tint GS, Shore V. Sitosterolemia. J Lipid Res. (1992) 33:945–55. doi: 10.1016/S0022-2275(20)41411-7
CrossRef Full Text | Google Scholar
- Gregg RE, Connor WE, Lin DS, Brewer HB Jr. Abnormal metabolism of shellfish sterols in a patient with sitosterolemia and xanthomatosis. J Clin Invest. (1986) 77:1864–72. doi: 10.1172/JCI112513
PubMed Abstract | CrossRef Full Text | Google Scholar
- Jamwal M, Aggarwal A, Maitra A, Sharma P, Bansal D, Trehan A, et al. First report of Mediterranean stomatocytosis/macrothrombocytopenia in an Indian family: a diagnostic dilemma. Pathology. (2017) 49:811–5. doi: 10.1016/j.pathol.2017.06.010
PubMed Abstract | CrossRef Full Text | Google Scholar
- Zhang ZQ, Chen AP, Yu T, Yang SJ, Yu DS, Yang R, et al. Exploring the pharmacological mechanism of danhe granules against hyperlipidemia by means of network pharmacology and verified by preliminary experiments. World J Tradit Chin Med. (2021) 7:436–44. doi: 10.4103/wjtcm.wjtcm_59_21
CrossRef Full Text | Google Scholar
- Pan Q, Zhang ZQ, Tian CY, Yu T, Yang R, Chai XL. Effect and signaling pathways of Nelumbinis folium in the treatment of hyperlipidemia assessed by network pharmacology. World J Tradit Chin Med. (2021) 7:445–55. doi: 10.4103/2311-8571.328619
CrossRef Full Text | Google Scholar
- Nguyen LB, Cobb M, Shefer S, Salen G, Ness GC, Tint GS. Regulation of cholesterol biosynthesis in sitosterolemia: effects of lovastatin, cholestyramine, and dietary sterol restriction. J Lipid Res. (1991) 32:1941–8. doi: 10.1016/S0022-2275(20)41897-8
PubMed Abstract | CrossRef Full Text | Google Scholar
Keywords: sitosterolemia, ABCG5 gene, xanthoma, surgery heterozygous pathogenic, therapy
Citation: Su S-Q, Xiong D-S, Ding X-M, Kuang J-A and Lin Y-C (2022) Pediatric patients with familially inherited sitosterolemia: Two case reports. Front. Cardiovasc. Med. 9:927267. doi: 10.3389/fcvm.2022.927267
Received: 24 April 2022; Accepted: 11 July 2022; Published: 16 August 2022.
Edited by:
Tomas Vaisar, University of Washington, United States
Reviewed by:
Nadezhda Sabeva, Central University of the Caribbean, Puerto Rico Babunageswararao Kanuri, The Ohio State University, United States
Copyright © 2022 Su, Xiong, Ding, Kuang and Lin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Di-Sheng Xiong, eGlvbmdkaXNoZW5neGRzQDEyNi5jb20=
